
Klinisk afprøvning af medicinsk udstyr
Anmeldelse
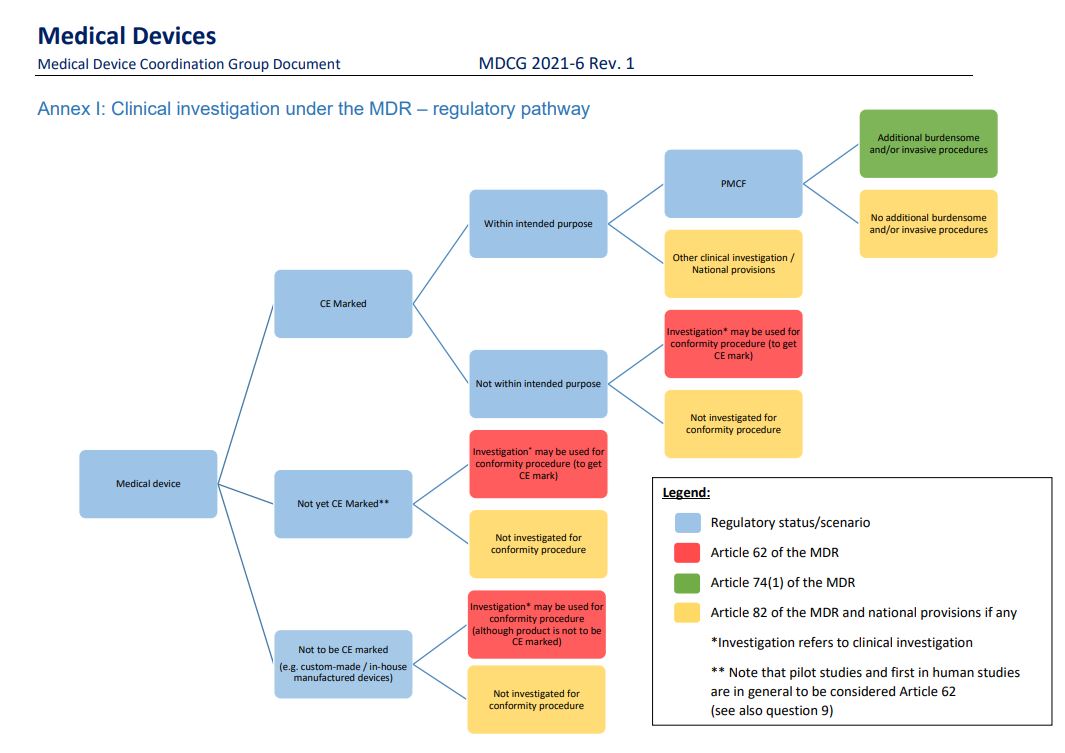
Anmeldelsespligten under de forskellige artikler i forordningen afhænger af, om udstyret er CE-mærket eller ej, og om der er intention om at anvende de indsamlede data til en overensstemmelsesvurdering af udstyret.Af figurerne nedenfor fremgår de forskellige typer af afprøvninger samt hvilke myndigheder de skal anmeldes til. Bemærk at kun for artikel 62 forsøg (markeret med rødt) er der krav om ekstern monitorering (f.eks. udført af GCP-enhederne), mens der for de øvrige kan være krav om intern monitorering
Der er 3 forskellige typer af kliniske afprøvninger af medicinsk udstyr angivet i MDR:
Artikel 62 ”Produktudvikling”: Afprøvning af medicinsk udstyr for at vurdere sikkerhed og ydeevne af udstyret, enten på ikke-CE-mærket medicinsk udstyr, eller på CE-mærket medicinsk udstyr, hvor fabrikanten ønsker at udvide produktets formål.
Artikel 74(1) ”PMCF-afprøvning med yderligere invasive/byrdefulde procedurer”: Fabrikantinitierede Post-Market kliniske opfølgningsstudier af CE-mærket udstyr, hvor forsøgsdeltagere udsættes for yderligere invasive/byrdefulde procedurer.
Artikel 82 ”Andre afprøvninger”.
Nedenstående figur er fra EU-Kommissionens vejledning ”MDCG 2021-6 Rev. 1 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation”.
Vejledningen findes her.
 Lægemiddelstyrelsen (LMST) og Videnskabsetiske Medicinske Komiteer (VMK) har gjort et samlet ansøgningsskema tilgængeligt. Sponsor skal indsende identiske ansøgninger til både LMST og VMK se link i højre side.
Lægemiddelstyrelsen (LMST) og Videnskabsetiske Medicinske Komiteer (VMK) har gjort et samlet ansøgningsskema tilgængeligt. Sponsor skal indsende identiske ansøgninger til både LMST og VMK se link i højre side. Anmeldelse af databehandling:
Se mere her vedrørende anmeldelse af databehandling.
Anmeldelse i offentligt tilgængeligt register
Inden inklusionsstart skal afprøvning med medicinsk udstyr anmeldes i et offentligt tilgængeligt register: clinicaltrials.gov. Se mere her.
Godkendelse til adgang til oplysninger fra patientjournaler
For at få adgang til oplysninger fra patientjournaler kræves der en godkendelse, det kan du læse mere om her.
Med forordningen for medicinsk udstyr, har EU Kommissionen tilgængeliggjort en database; Eudamed. Eudamed indsamler og behandler oplysninger om medicinsk udstyr på markedet og gør oplysningerne tilgængelige for offentligheden. I Q4 2027 forventes det at modulet i databasen til anmeldelse af klinisk afprøvning med medicinsk udstyr skal anvendes.
Ændringer i projektet
Anmeldelse af ændringer i den kliniske afprøvning sker på to forskellige måder alt efter om forsøget fik tilladelse inden den 26.05.2021 eller senere. Hvis der er givet tilladelse inden den 26.05.2021 anvendes det relevante skema (se link i højre side), som sendes til LMST og til den Regionale Videnskabsetiske Komité, der i sin tid vurderede projektet.For kliniske afprøvninger, der har fået tilladelse efter den 26.05.2021, skal ændringen ansøges samtidigt hos LMST og VMK på (se link i højre side). Skemaet sendes til de to myndigheder samtidigt - mail oplysninger fremgår af skemaet.