Elektroniske systemer
Det er i dag næsten umuligt at gennemføre kliniske forsøg uden brug af elektroniske systemer. Det kan både være systemer, der anvendes i den daglige kliniske praksis (fx journalsystemer, scannere, CGM), eller systemer, der er designet eller tilrettet generelt til kliniske forsøg (fx eCRF, ePRO, AI-algoritmer).
Med den stigende anvendelse af teknologi i kliniske forsøg er det afgørende at have klarhed over, hvem der er ansvarlig for de enkelte elektroniske systemer, samt at sikre, at systemerne er validerede og egnede til formålet (”fit for purpose”).
Både ICH-GCP E6 (R3) og ”
Guideline on computerised systems and electronic data in clinical trials” lægger vægt på, at elektroniske systemer skal sikre at kliniske data er pålidelige, sporbare, beskyttede og af høj kvalitet – og at teknologien bruges på en måde, der beskytter forsøgsdeltagerne og opfylder GCP-principperne.
Det betyder, at alle systemer, der håndterer data skal kunne dokumentere:
1) Dataintegritet og ALCOA
Dataintegritet (se ALCOA) er et grundprincip i GCP og skal sikres gennem hele datalivscyklussen, fra oprettelse, indsamling, arkivering til sletning.
Metadata (fx audit trails), er en integreret del af systemet og er med til at dokumentere oprindelse og kontekst. Ingen data er komplette uden deres tilhørende metadata.
2) Sikkerhed og sporbarhed
Sporbarhed skal være fuldstændig (audit-trail) — alle data og ændringer i data skal kunne spores til en unik bruger, tidspunkt og hvis årsag til ændringen ikke er tydelig, bør der være mulighed for at give en begrundelse.
Dokumentation af dataflow skal foreligge for at opretholde sporbarhed.
Systemet skal beskytte data mod tab, manipulation og uautoriseret adgang.
Dokumentation for test af backup/restore.
Elektroniske overførsler skal ske sikkert.
Data skal være arkiverbare og tilgængelige i hele opbevaringsperioden.
Hvis systemet bliver udfaset, skal data stadig kunne hentes og læses (migrering kan blive nødvendig).
3) Adgangskontrol og beskyttelse af data
Adgang skal være baseret på roller og behov (strictly ”need to know”) og der skal løbende føres log over tildelte og ophævede brugeradgange. Log bør vise tidspunkter for oprettelse, ophævelse af brugerkonti, ændringer i bruger-roller, tilladelser og adgange.
Systemer skal forhindre, at brugere får adgang til funktioner eller data, de ikke er autoriseret til.
Data skal beskyttes mod uautoriseret ændring, tab eller afsløring — både teknisk og organisatorisk.
4) Validering og ændringsstyring
Systemer skal inden de tages i brug være validerede til formålet.
Der skal foreligge dokumentation for validering/tests, som bevis for, at systemet fungerer som tiltænkt.
Ændringer i systemet kræver ændringsstyring (change control) og evt. gentaget validering.
Ansvaret for ovenstående påhviler enten sponsor eller investigator – også selvom der benyttes ”service providers”.
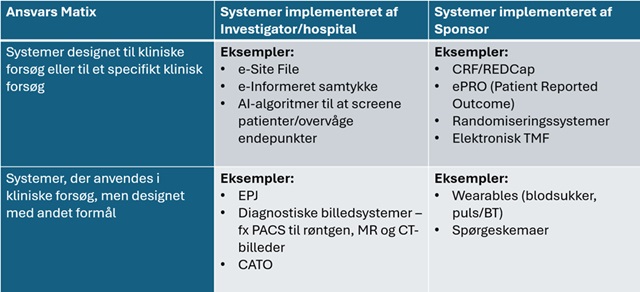
Roller og ansvar
Ansvar for elektroniske systemer kan overordnet opdeles efter:
Om systemet er implementeret af sponsor eller investigator
Om systemet er designet til kliniske forsøg eller anvendes i den almindelige kliniske praksis
Afhængigt af systemtypen kan ansvaret derfor variere.
 Sponsor
Sponsor har det overordnede ansvar for, at elektroniske systemer og datahåndtering opfylder kravene i ICH-GCP E6(R3) - og at brugere er tilstrækkeligt trænede. Dette indebærer, at sponsor ud fra en risikobaseret tilgang sikrer, at der for systemer designet til det specifikke kliniske forsøg:
Føres log over anvendte elektroniske systemer, samt opbevarer dokumentation for hvert systems funktionalitet, grænseflader (interfaces) og valideringsstatus.
Sikres at hvert system er tilstrækkelig vedligeholdt mht. interne og eksterne sikkerhedsforanstaltninger, validering, audit trail, backup, restore, IT-sikkerhed, mv.
Sikre at der er en kommunikationsvej imellem eventuelle service providers og sponsor/investigator mht. systemnedbrud/fejl.
Føre log over adgange til systemet.
- Sikre at adgangstilladelser til projektpersonale på sites er i overensstemmelse med investigators uddelegeringer, samt at disse adgangstilladelser er synlige for investigator.
- Sikre træning af brugere.
For systemer, der
ikke er designet specifikt til forsøget, men indeholder kildedata (f.eks. Patientjournalen), bør sponsor sikre at der foreligger dokumentation for, at systemerne anses for egnede, i relation til brug af data i kliniske forsøg.
Investigator skal sikre korrekt brug, adgang og dataindtastning på sites jf. godkendt protokol og andre forsøgsrelateret dokumenter. Investigator er ansvarlig for elektroniske systemer der tages i brug på eget site specifikt til forsøget. Det indebærer at investigator foretager ”fit for purpose” vurdering og relevant træning af projektpersonale i brug af systemet.
Investigator skal ligeledes sikre, at ovenstående generelle krav (beskrevet i pkt. 1-4) til elektroniske systemer er opretholdt og dokumenteret. Hvor der udleveres elektroniske systemer (fx wearables) til projektdeltagere, skal investigator sikre, at denne udlevering trackes og at deltageren er trænet tilstrækkeligt.
